MS Amanda is a scoring system to identify peptides out of tandem mass spectrometry data using a database of known proteins.
This algorithm is especially designed for high resolution and high accuracy tandem mass spectra. One advantage of MS Amanda is the high speed of spectrum identification, especially since MS Amanda 2.0. In addition, MS Amanda is also very accurate, as we observe a high overlap of identified spectra with gold-standard algorithms Mascot and SEQUEST.
Since MS Amanda 3.0 automatic validation using Percolator can be performed.
More information about the implementation of MS Amanda and how the MS Amanda algorithm works is given here:
Please download the latest version of MS Amanda from the The MS Amanda GitHub repository
In addition, MS Amanda Standalone is also integrated in SearchGUI and PeptideShaker!
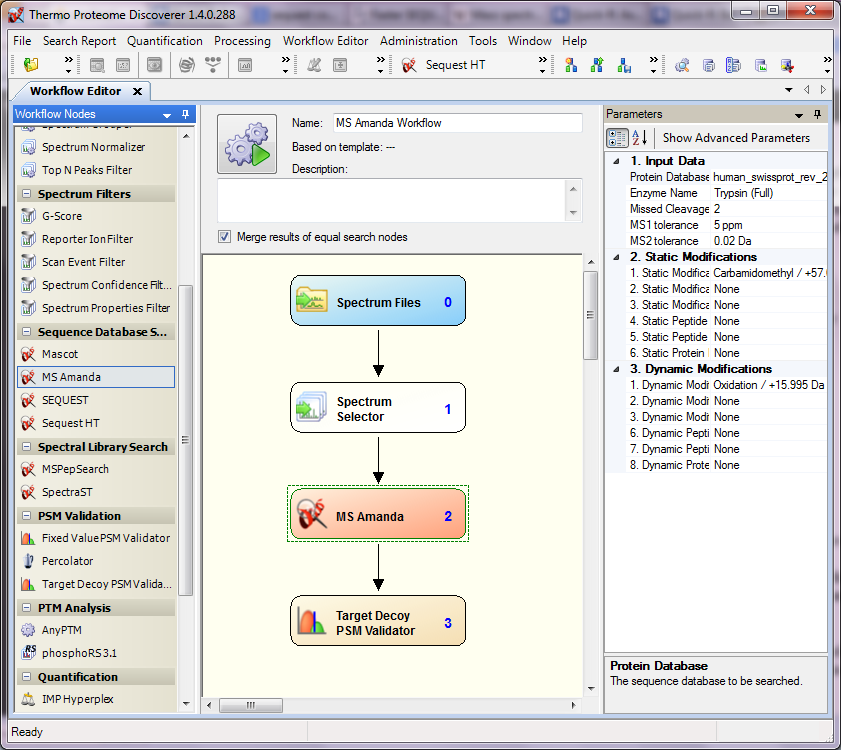
A detailed description on how to use MS Amanda for Proteome Discoverer, on the parameters provided by MS Amanda, and how to filter results obtained with MS Amanda can be found in the MS Amanda Manual for PD.
A typical MS Amanda PD workflow can be found here.
This research project is a collaboration of the Protein Chemistry Group at IMP and the Bioinformatics Research Group at FH Upper Austria, Hagenberg Campus.
For any further questions, bug reports, ideas,... please contact
Viktoria Dorfer,
Marina Strobl,
Stephan Winkler, or
Karl Mechtler, or
open an issue in the The MS Amanda GitHub repository,
or post your comment in the MS Amanda Google Group.



{kind=link}