IMP-apQuant is a Proteome Discoverer node for accurate and precise label-free quantification of peptides and proteins.
This sample workflow should make you ready to go. It is the workflow to analyse the Ramus et al data set which is also used in the performance comparison further down in the Performance section.
We have a forum. Feel free to ask questions and give feedback!
We benchmarked IMP-apQuant on a publicly available dataset (Ramus et al). The original raw files are available on PRIDE (PXD001819). The samples are comprised of UPS1 protein mix spiked in at different concentrations into a 1:1 yeast background. This dataset was used in a comparison for different LFQ algorithms (Veit et al). We used results published there as Supplementary Data in order to compare the performance of IMP-apQuant to LFQProfiler(OpenMS), MaxQuant, MFPaQ and Skyline.
IMP-apQuant outperforms these tools by enabling the detection of more differentially abundant UPS proteins at the same number of falsely identified (unregulated) yeast proteins.
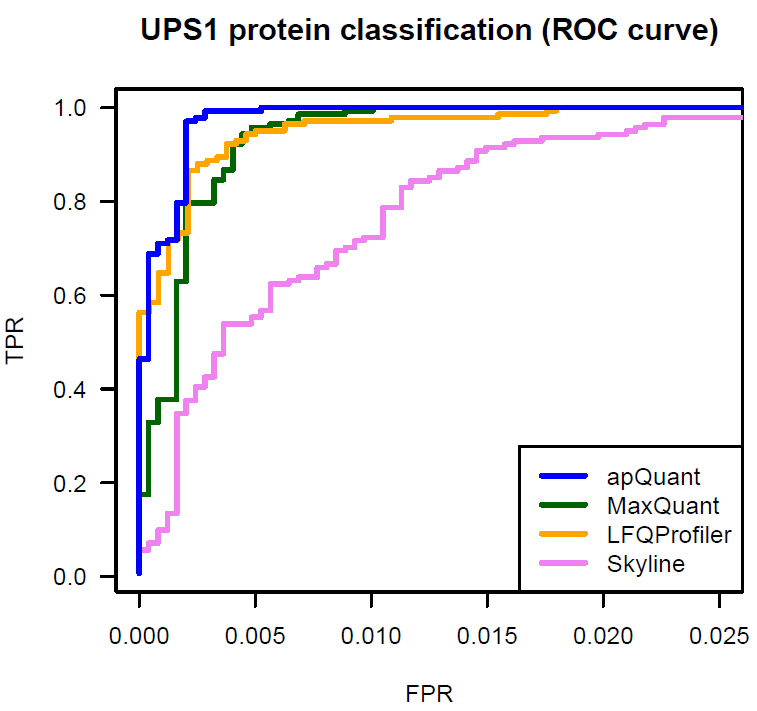
The Proteome Discoverer workflow to reproduce this result is available for download and provides a starting point for easy incorporation of IMP-apQuant in your workflow. Furthermore the final results file is also available here.
| Date | Version | Info |
|---|---|---|
| 2019-01-28 | 3.1 | included crosslink quantification |
| 2017-12-19 | 3.0 | project renamed to IMP-apQuant |
| 2017-05-30 | 3.0 | feature based scoring of quantification confidence, iBAQ support |
| 2016-11-09 | 2.18 | RAM and time usage reduced, stability fixes |
| 2016-07-25 | 2.15 | scoring finalized, stability fixes |
| 2016-06-16 | 2.14 | RAM usage decreased, separate installer for PD 2.0 |
| 2016-06-02 | 2.13 | several small bug fixes |
| 2015-05-25 | initial release |
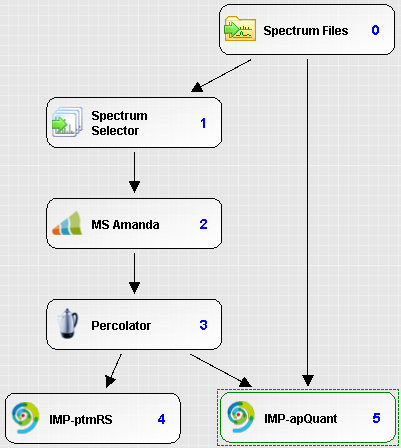
Processing workflow
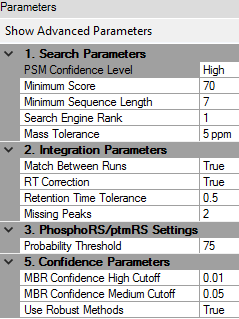
Processing parameters

Consensus workflow

Consensus parameters
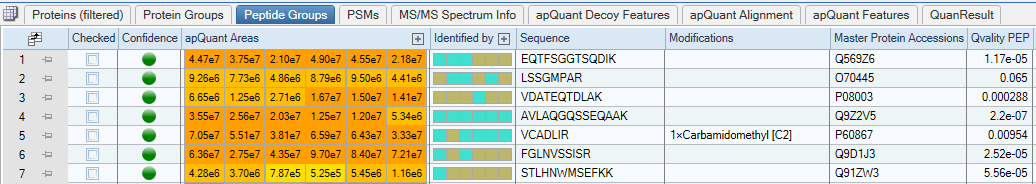
Peptides

Proteins
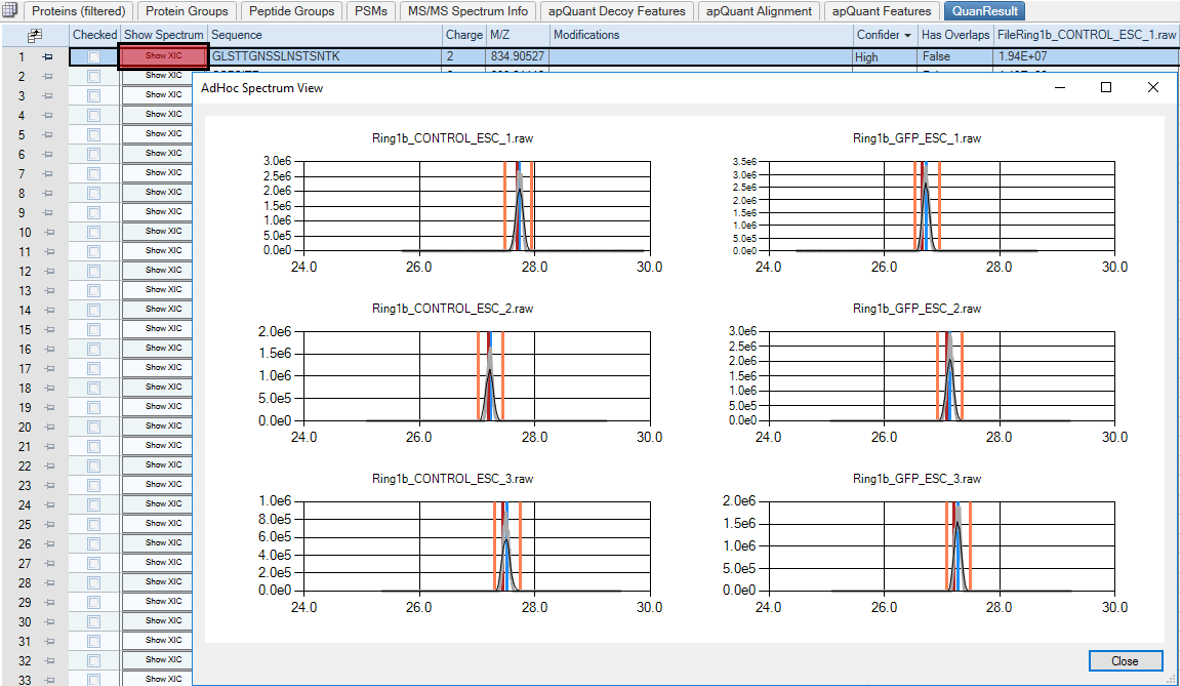
Extracted ion chromatogram of a peptide in six samples.
PSMs are depicted in red, integration boundaries in orange and the peaks center of gravity in blue.


