phosphoRS v3.1
This tool enables automated and confident localization of modification sites within validated peptide sequences and can be applied to all commonly used fragmentation techniques (CID, ETD and HCD). It calculates individual probability values for each putatively modified site based on the given MS/MS data. For further information on the algorithm of the software, please refer to the corresponding publication.
What's new?
The phosphoRS 3.1 node is the latest version of this phospho-site localization algorithm and is compatible with both Thermo Scientific Proteome Discoverer 1.3 and 1.4. The new features of this update are:
- Multithreading
- phosphoRS 3.1 automatically adapts its processing performance to the individual system resources.
- Direct access
- The actual phosphoRS site localization algorithm can now be accessed directly via a DLL, whithout requiring the installation of Thermo Scientific Proteome Discoverer. This enables you to integrate phosphoRS into your individual workflow. See the Design section for detailled information.
- Individual peak depth approach
- The optimal number of peaks being considered (i.e. the best peak depth) for localization of phosphorylation sites is determined for each m/z window individually, which increases the sensitivity of site localization for CID data.
- Optimized scoring parameters
- Depending on the applied fragmentation technique different fragment ion types are used for scoring in order to provide highest sensitivity. For CID data, only singly and doubly charged b and y ions are scored, while for analysis of HCD spectra also neutral loss ions are considered. In contrast, when localizing phosphorylation sites in ETD spectra, exclusively singly charged c, z and z+H ions are taken into account.
- Additional node parameters
- The phosphoRS node allows the user to specify for example if neutral loss peaks should be considered for scoring (true: always consider them, false: never consider them, automatic: neutral loss peaks are considered only for HCD data). Moreover, one can set the maximum number of modification-isoforms and PTMs per peptide that should be considered. If a certain peptide exceeds this cutoff it will not be analyzed by the node.
- Altered output column headings and visibilities
- In order to simplify the output of phosphoRS corresponding column headings in Thermo Scientific Proteome Discoverer have been changed. Further, the binomial peptide score column and the isoform confidence probability column are hidden by default.
Further we provide an easy-to-use Microsoft Excel macro, which facilitates the analysis of your phosphoRS 3.1 node results. First, the filtered results have to be exported from Thermo Scientific Proteome Discoverer to Microsoft Excel and then the data can be copied to the downloadable template for further processing.

Performance
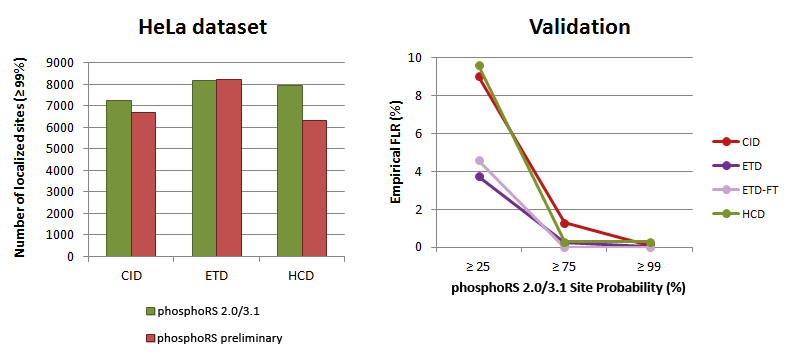
The analysis of a HeLa dataset revealed that phosphoRS 3.1 is able to localize more phosphorylation sites compared to the preliminary version (distributed with Thermo Scientific Proteome Discoverer 1.3), which is attributed to the novel individual peak depth approach and the optimization of scoring parameters. Repeating the validation of the algorithm by analyzing chemically synthesized phospho-peptides with precisely known sites ensured that the empirical false localization rate (FLR) is always lower than indicated by the applied phosphoRS site probability cutoff. All results are identical between phosphoRS 2.0 and 3.1.
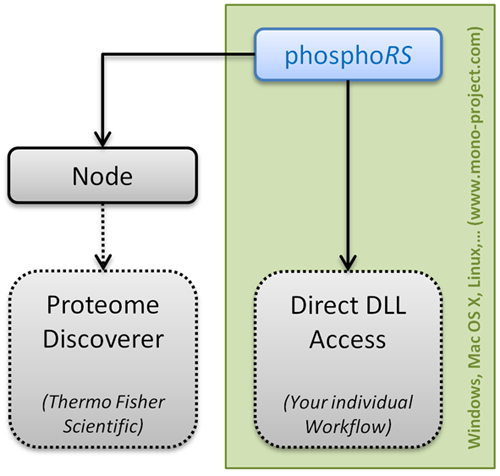
Design
As previously mentioned, the phosphoRS 3.1 code has been seperated into two DLLs, one containing the actual site localization functionality (IMP.PhosphoRS.dll) and the other being the interface to Thermo Scientific Proteome Discoverer (IMP.PhosphoRS2Nodes.dll). This new software architecture allows bioinformaticians now to integrate phosphoRS into their individual data analysis workflows. We provide a simple demo showing how to access the complete functionality of phosphoRS here. Please do not hesitate to contact us in case of questions regarding phosphoRS.
Download & Installation
The phosphoRS 3.1 node can be installed easily for Thermo Scientific Proteome Discoverer 1.3/1.4 and will replace its preliminary version by following these steps:
- Download the node's DLLs for Thermo Scientific Proteome Discoverer 1.3 or 1.4 and read carefully the attached license agreement. Proceed only if you agree to the terms and conditions of the license.
- Save all your files and shutdown Thermo Scientific Proteome Discoverer.
- Navigate to the folder where you have installed Thermo Scientific Proteome Discoverer (Tip: You can easily find out the path by right-clicking the Thermo Scientific Proteome Discoverer desktop icon and open the Properties window. The folder path is written in the field Target.)
- Move the old phosphoRS DLLs to a backup folder and copy the new ones into the Thermo Scientific Proteome Discoverer folder.
-
Unblock both DLLs if required by right-clicking each DLL, opening its properties window and clicking the Unblock button if available.
-
Restart Thermo Scientific Proteome Discoverer, navigate to the licensing page and click on Scan for Missing Features. Subsequently, restart the program once more.
-
If the update to phosphoRS 3.1 was successful, then the name of the node will change to phosphoRS3.1
-
Additionally, you will be provided with the following (advanced) node parameters.


